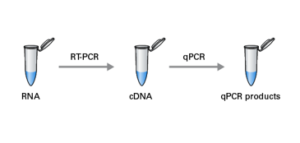
To avoid amplification of genomic DNA in total RNA samples:
- Design primers that avoid genomic amplification: select a large intron region and design forward and reverse primers in exons upstream and downstream of the intron. With this strategy, genomic amplifications cannot occur for large introns. When introns are small, genomic amplification can be differentiated based on differences in melting temperature as the result of different amplification sizes (melt curve).
- For genes that lack introns or when the genomic structure is unknown, treat the total RNA with DNase I to remove genomic DNA.
Target gene expression levels are often normalized to the expression of a “housekeeping gene” (reference gene) to correct for differences in the amount of input RNA and variations in reaction efficiency. How do I select a suitable housekeeping gene?
There is no single, universally appropriate housekeeping gene suitable for accurate normalization in every experimental condition, as it is important to select housekeeping genes that do not vary in expression levels in the experimental system used. Common housekeeping genes that have been used in the past include GAPDH and β-actin; however, reports in recent years indicate that expression of these genes may also vary, depending on the sample type and/or experimental conditions.
Using multiple housekeeping genes for normalization is currently the most reliable approach. In this strategy, the expression of multiple housekeeping genes is assayed, and the genes that show the lowest level of variation are selected for use. Software has been developed for selecting the optimum genes for correction (e.g., geNorm and BestKeeper). Inferences may also be made from microarray or RNA-seq expression profiling data, if available.